
第一類醫療器械實行產品備案管理,第二類、第三類醫療器械實行產品注冊管理。向我國境內出口第二類、第三類醫療器械的境外生產企業,應當由其在我國境內設立的代表機構或者指定我國境內的企業法人作為代理人,向國務院食品藥品監督管理部門提交注冊申請資料和注冊申請人所在國(地區)主管部門準許該醫療器械上市銷售的證明文件。第二類、第三類醫療器械產品注冊申請資料中的產品檢驗報告應當是醫療器械檢驗機構出具的檢驗報告;臨床評價資料應當包括臨床試驗報告,但依照本條例第十七條的規定免于進行臨床試驗的醫療器械除外。...
第一類醫療器械實行產品備案管理,第二類、第三類醫療器械實行產品注冊管理。向我國境內出口第二類、第三類醫療器械的境外生產企業,應當由其在我國境內設立的代表機構或者指定我國境內的企業法人作為代理人,向國務院食品藥品監督管理部門提交注冊申請資料和注冊申請人所在國(地區)主管部門準許該醫療器械上市銷售的證明文件。第二類、第三類醫療器械產品注冊申請資料中的產品檢驗報告應當是醫療器械檢驗機構出具的檢驗報告;臨床評價資料應當包括臨床試驗報告,但依照本條例第十七條的規定免于進行臨床試驗的醫療器械除外。
1.申請表
2.證明性文件
3.醫療器械安全有效基本要求清單
4.綜述資料
5.研究資料
6.生產制造信息
7.臨床評價資料
8.產品風險分析資料
9.產品技術要求
10.產品注冊檢驗報告
11.說明書和標簽樣稿
12.符合性聲明
1.受理
申請人向國家藥品監督管理局行政受理服務大廳提出申請,受理人員根據申報事項按照《國家食品藥品監督管理總局關于公布醫療器械注冊申報資料要求和批準證明文件格式的公告》(2014年第43號)、《食品藥品監管總局關于印發境內第三類和進口醫療器械注冊審批操作規范》(食藥監械管〔2014〕208號)的要求對申報資料進行形式審查。
申請事項屬于本部門職權范圍,申報資料齊全、符合形式審查要求的,予以受理;申報資料存在可以當場更正的錯誤的,允許申請人當場更正;申報資料不齊全或者不符合形式審查要求的,在5個工作日內一次告知申請人需要補正的全部內容,逾期不告知的,自收到申報資料之日起即為受理;申請事項不屬于本部門職權范圍的,即時告知申請人不予受理。
2.審查
受理人員自受理之日起3個工作日內將申報資料轉交技術審評機構。
技術審評機構應當在60個工作日內完成第二類醫療器械注冊的技術審評工作。需要外聘專家審評、藥械組合產品需與藥品審評機構聯合審評的,所需時間不計算在內,技術審評機構應當將所需時間書面告知申請人。技術審評過程中需要申請人補正資料的,技術審評機構應當一次告知需要補正的全部內容。申請人應當在1年內按照補正通知的要求一次提供補充資料。技術審評機構應當自收到補充資料之日起60個工作日內完成技術審評。質量管理體系核查的時間和申請人補充資料的時間,不計算在審評時限內。
3.許可決定
國家藥品監督管理局應當在技術審評結束后20個工作日內作出決定,對符合安全、有效要求的,準予注冊。對不予注冊的,應當書面說明理由,并同時告知申請人享有申請復審和依法申請行政復議或者提起行政訴訟的權利。
4.送達
自作出審批決定之日起10個工作日內,國家藥品監督管理局行政事項受理服務和投訴舉報中心將行政許可決定送達申請人。
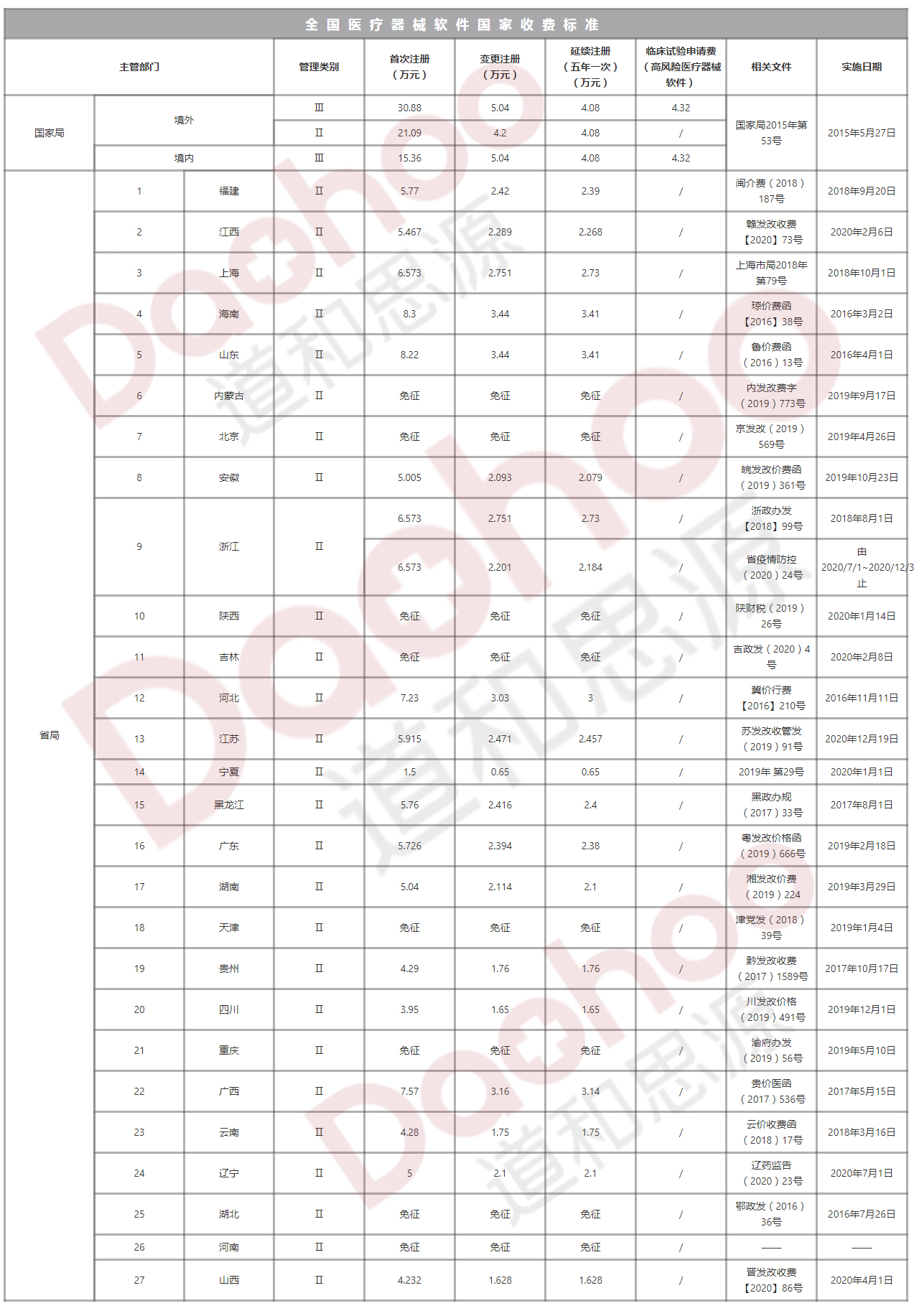
進口第二類醫療器械首次注冊費:21.09萬元
國家發展改革委《關于重新發布中央管理的食品藥品監督管理部門行政事業性收費項目的通知》(財稅〔2015〕2號)和《關于印發〈藥品、醫療器械產品注冊收費標準管理辦法〉的通知》(發改價格〔2015〕1006號),《國家食品藥品監督管理總局關于發布藥品、醫療器械產品注冊收費標準的公告》(2015年第53號)。
進口產品提交申報資料有哪些要求?
答:依據《境內第三類和進口醫療器械注冊審批操作規范》(食藥監械管[2014]208號)的要求,進口產品申報資料,如無特別說明,原文資料均應由申請人簽章,中文資料由代理人簽章。原文資料“簽章”是指:申請人法定代表人或者負責人簽名,或者簽名并加蓋組織機構印章,并且應當提交由申請人所在地公證機構出具的公證件。

 京公網安備 11010502043733號??京ICP備16013397號-1??XML地圖 ?網站地圖?熱門TAG?2018最新《醫療器械分類目錄》
京公網安備 11010502043733號??京ICP備16013397號-1??XML地圖 ?網站地圖?熱門TAG?2018最新《醫療器械分類目錄》









